SYBR Green I | Real-Time PCR and Melting Analysis
The first LightCycler real-time curves used SYBR Green I to monitor target amplification. At that time (early in 1995), SYBR Green I was a dye just introduced by Molecular Probes for gel analysis of DNA. It had not been used for real-time PCR and was thought to be heat labile. However, its spectral properties (close to fluorescein) and high specificity to double-stranded DNA were much better than ethidium bromide. Contrary to the bias at the time, SYBR Green I worked very well for real-time PCR, and has become the most popular method of real-time monitoring for simple reasons of cost and convenience. Synthesis and labeling of probes became unnecessary. When SYBR Green I fluorescence is monitored once each cycle, the initial template concentration can be quantified. However, low concentrations of target can be difficult to quantify because unintended products may also be amplified and detected. These specificity concerns are not as important today because of hot-start methods and the common use of melting curve analysis to identify products.
Hybridization Probes
Our initial attempts in 1994 to work with hydrolysis probes (TaqMan probes) failed, so we developed an alternative probe system that later was launched with the LightCycler. The signal from these adjacent hybridization probes (HyProbe, FRET probes, or kissing probes) depends directly on hybridization, not on exonuclease activity and probe hydrolysis. Each probe is covalently labeled with only one dye, so they are inherently simpler to synthesize than double-labeled probes. One probe is labeled on the 3'-end. The other probe is labeled on the 5'-end and its 3'-end blocked to prevent extension. When both probes are hybridized, fluorescence resonance energy transfer occurs. Maximum fluorescence occurs with a one base separation between probes. Hybridization probes are commonly used today for quantification and genotyping when high specificity is required.
The fluorescence signal is even more specific than single probe systems because hybridization of 4 oligonucleotides (2 primers and 2 probes) is required. Similar to SYBR Green I, fluorescence can be monitored once each cycle for quantification, or continuously to assess hybridization. Hybridization probes are used in the FDA approved kits for factor V Leiden and prothrombin mutations, and in Roche's Septifast system for identifying the causative agents of septicemia. Hybridization probes can be multiplexed by melting temperature, fluorescence color, or by a combination of both.
Real-Time Quantification
SYBR Green I and fluorescent probes can be used in real-time PCR for template DNA quantification. Fluorescence from each sample is collected once each cycle during PCR, and plotted against cycle number. The starting template concentration correlates inversely to the time of first appearance of fluorescence signal. Signal appears earlier (at lower cycle number) the higher the concentration of template. Because PCR is exponential, the correlation is logarithmic. Specifically, the logarithm of the starting template concentration is inversely proportional to the fractional cycle number that identifies the horizontal position of the real-time amplification curve. Absolute quantification is possible by comparing an unknown to a standard curve, as long as the efficiency of amplification is constant.
The fractional cycle number can be estimated in different ways, often requiring baseline estimation and adjustment. A baseline-independent method to determine the fractional cycle number was introduced with the LightCycler as the second derivative maximum of each curve. The second derivative maximum method is not affected by baseline artifacts and allows more precise quantification. The fractional cycle number (originally defined by Higuchi as the threshold cycle Ct) is referred to as the crossing point (Cp) on the LightCycler. Often, absolute quantification is not required. In this case, standards and standard curves are not necessary. Instead, the relative quantity of target can be obtained by the difference in fractional cycle numbers between samples. Results can be normalized to a reference or "housekeeping" target if necessary. Although real-time PCR is highly sensitive, it has stochastic limitations. When the concentration of target is too low, random chance dictates how many copies end up in a PCR tube. The probability of ending up with no template in a PCR is about 1% when the average number of copies per PCR is five. This random variation affects precision at low copy numbers of target. Relative quantification of different alleles or pseudogenes is possible by melting analysis after competitive PCR.
Genotyping by Melting Analysis
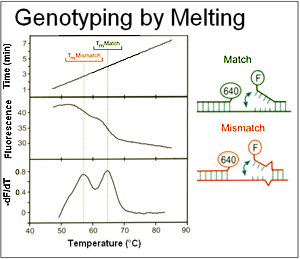
Hybridization probes were initially developed for quantification in the LightCycler. However, the possibility of using these probes to detect different alleles was attractive. Instead of "static" genotyping by incubating an immobilized probe:target hybrid at a single temperature, we were able to watch the melting process in solution as it occurred by monitoring fluorescence as the sample was heated. This work, originally published in 1997 with factor V Leiden, was rapidly extended to small insertions/deletions (e.g., F508del of the CFTR gene) and many other targets. Because different variants under the probe result in different probe stabilities, different alleles can be distinguished by their Tms.
Multiplexing by color and temperature allowed higher level multiplexing. Later, we found out that in many cases, only one probe was necessary. Fluorescein-labeled probes are often quenched or de-quenched by hybridization. This effect depends on the sequence surrounding the fluorescein label, particularly the presence of G bases. An additional probe modification resulted in stable de-quenching upon hybridization, and is trademarked as SimpleProbe by Roche.
Genotyping with Probes
Many genotyping methods that rely on melting were developed in conjunction with the LightCycler. Although hydrolysis probes (TaqMan) are commercially prevalent today, hybridization probes include dual hybridization probes (Wittwer et al. 1997), single hybridization probes (Crockett & Wittwer, 2001), unlabeled probes (Zhou et al. 2004), and snapback primers (Zhou et al. 2008). Dual hybridization probes are used for quantification and genotyping, while single hybridization probes, unlabeled probes, and snapback primers are primarily used for genotyping. While hydrolysis probes typically require 1 probe for each allele, hybridization probes can often identity several alleles, depending on the instrument resolution. Over time hybridization probes became less complex, requiring fewer probes, covalent labels, and oligonucleotides for amplification. While dual hybridization probes required 2 probes each with a single label, single hybridization probes used only one probe with one label, unlabeled probes had no covalent label, and snapback primers incorporated an unlabeled probe into a primer tail so that only 2 primers were necessary for genotyping.
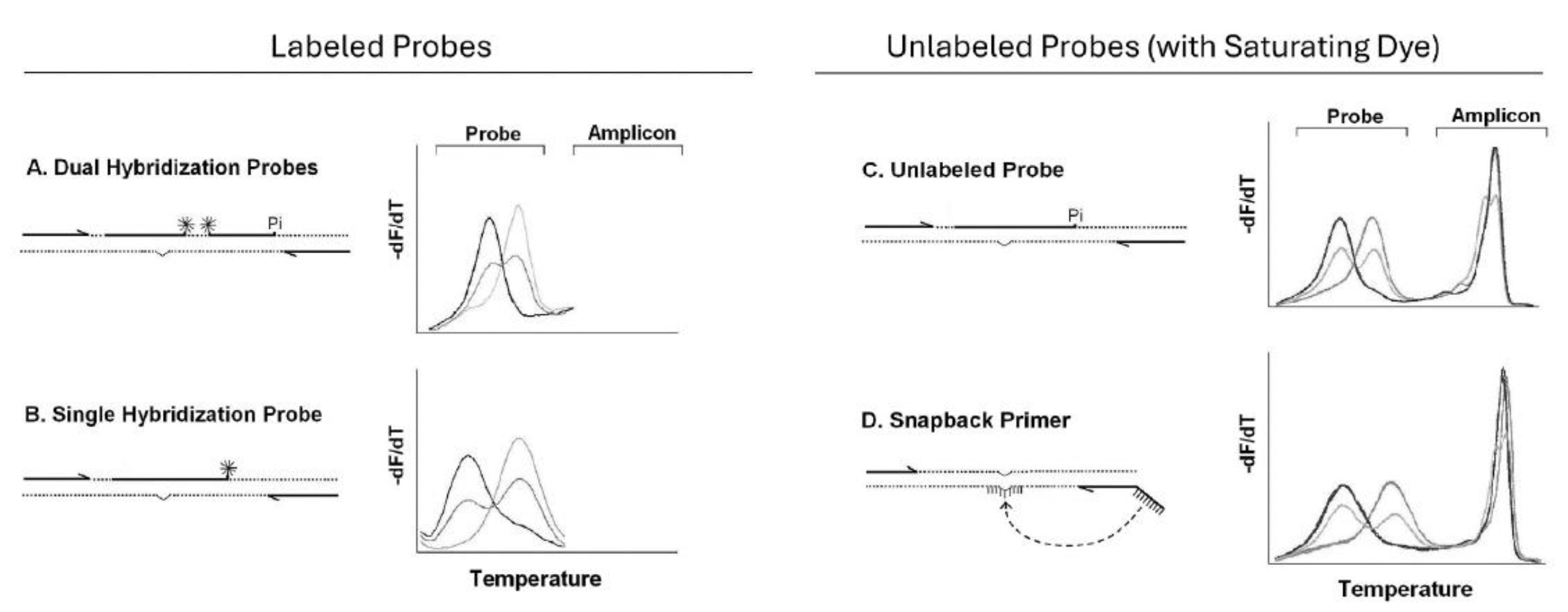
Unlabeled probes and snapback primers perform best with saturating dyes and high resolution melting instruments that track both probe and amplicon transitions. Unlike hydrolysis probes, fluorescence is monitored over a range of temperatures, rather than at a single temperature. Different alleles melt at different temperatures, and heterozygotes are easy to distinguish from homozygotes by a double peak on negative derivative melting curve plots.
Innovation Beyond the LightCycler
The LightCycler continues to influence innovation in our lab. Click on the topics below for more information!